
 澳大利亚
澳大利亚 韩国
韩国 巴西
巴西 日本
日本 俄罗斯
俄罗斯 中国台湾
中国台湾 中国
中国 中国香港
中国香港 欧盟
欧盟 印度尼西亚
印度尼西亚 马来西亚
马来西亚 新加坡
新加坡 菲律宾
菲律宾 美国
美国 加拿大
加拿大 印度
印度 越南
越南 泰国
泰国 沙特阿拉伯
沙特阿拉伯日本PMDA医疗器械临床评价报告的要求及建议(一)

今天,我们的讨论与日本医疗器械的临床相关。我们举目望去,目前境外诸国被新冠困扰,在疫情严重的非常时刻,让我们静下心,学习一些有价值的内容,提升大家的价值,真正做到为将来做准备。相信今天的讨论,有一定的价值。
我们知道在日本,医疗器械想上市,一般有三条路:
1) I类备案;
2) II/III(部分)类RCB(Recognized Certificate Body)审核;
3) III/IV 类PMDA(独立行政法人机构)审核。
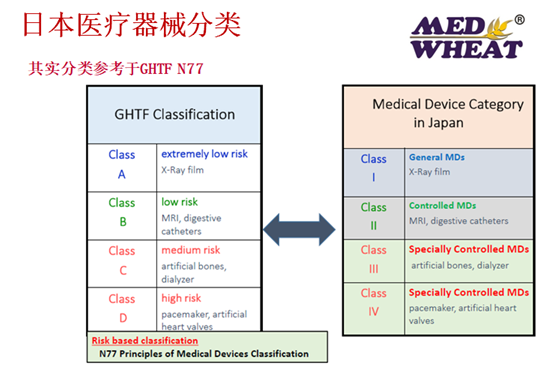
日本的医疗器械分类一般遵循GHTF的分类明细,所以大家在产品往日本注册前,可以通过查询GHTF的分类要求或查询日本医疗器械数据库,来获得具体的分类确认。一般在绝大部分情况下,我们中国的医疗器械耗材类,不少都是属于I、II、III类,很多都可以通过备案和RCB审核,然后MAH持证。其实日本还有一种器械分类的方法,如下几种情况:
1) 创新型医疗器械(NovelDevice)- 需要提供临床历史数据;
2) 改良型医疗器械(ImprovedDevice)且无审核基准 - 需要提供临床历史数据;
3) 相同型医疗器械(Me-tooDevice)且无审核基准 - 需要提供临床历史数据。
一般通过PMDA审核的器械,或多或少会有相关临床数据的考量,需要提供“临床评价报告”(Clinical Evaluation Report)。“临床评价报告”可以认为与日本注册申请所需的临床研究试验结果有相同评估的作用,不过需要注意这里提到的临床评价的范围比欧盟定义的“临床评价”范围要窄,欧盟针对医疗器械的整个生命周期的,而日本的临床报告一般针对的是该器械上市前的数据。
日本《药品和医疗器械法实施条例》中,第274条规定的医疗器械规定,对于那些被要求必须提交临床试验相关资料的医疗器械,比如创新医疗器械和改良医疗器械(需要临床数据才可以申请的),当仅使用现有临床数据而不进行新的临床试验就可以评估临床有效性和安全性时,但是批准申请书随附的临床评估报告不受GCP合格性调查的约束,但可能受制于申请材料的可靠性标准相符的准则《实施条例第114-22条》。
无基准的中高风险器械,在日本都需要交临床评价报告,总结并评价上市前临床历史数据,在数据充分性和可靠性的基础上,PMDA可能可以豁免真实临床或批准小规模临床。日本PMDA接受符合境外GLP的上市前临床数据。
(未完待续)
往期回顾:日本PMDA审核机构介绍
