
 澳大利亚
澳大利亚 韩国
韩国 巴西
巴西 日本
日本 俄罗斯
俄罗斯 中国台湾
中国台湾 中国
中国 中国香港
中国香港 欧盟
欧盟 印度尼西亚
印度尼西亚 马来西亚
马来西亚 新加坡
新加坡 菲律宾
菲律宾 美国
美国 加拿大
加拿大 印度
印度 越南
越南 泰国
泰国 沙特阿拉伯
沙特阿拉伯日本PMDA审核机构介绍

一、PMDA 概述
PMDA 全称为Pharmaceuticalsand Medical Devices Agency,其日语名称翻译为“独立行政法人医药品医疗器械综合机构”, 是厚生劳动省医药食品局所管辖的独立行政法人。
PMDA是2004年4月1日成立的,它的前身由国家卫生科学研究所the National Institute of Health Sciences、日本医疗设备促进会the Japan Association for the Advancement of Medical Equipment Center(JAAME)、以及药品安全与研究组织theOrganization for Pharmaceutical Safety and Research(OPSR)的一部分组成。(见图1)

PMDA 的业务主要包括审查、安全对策、健康损害救济三大板块,如下(见图2):
a) 提供批准(批准审查)和收集;
b) 分析和提供上市后安全信息(安全措施)的指导和审查;
c) 控制不良时间,为改善国民健康做出贡献。

PMDA 2019年的组织机构见图3。
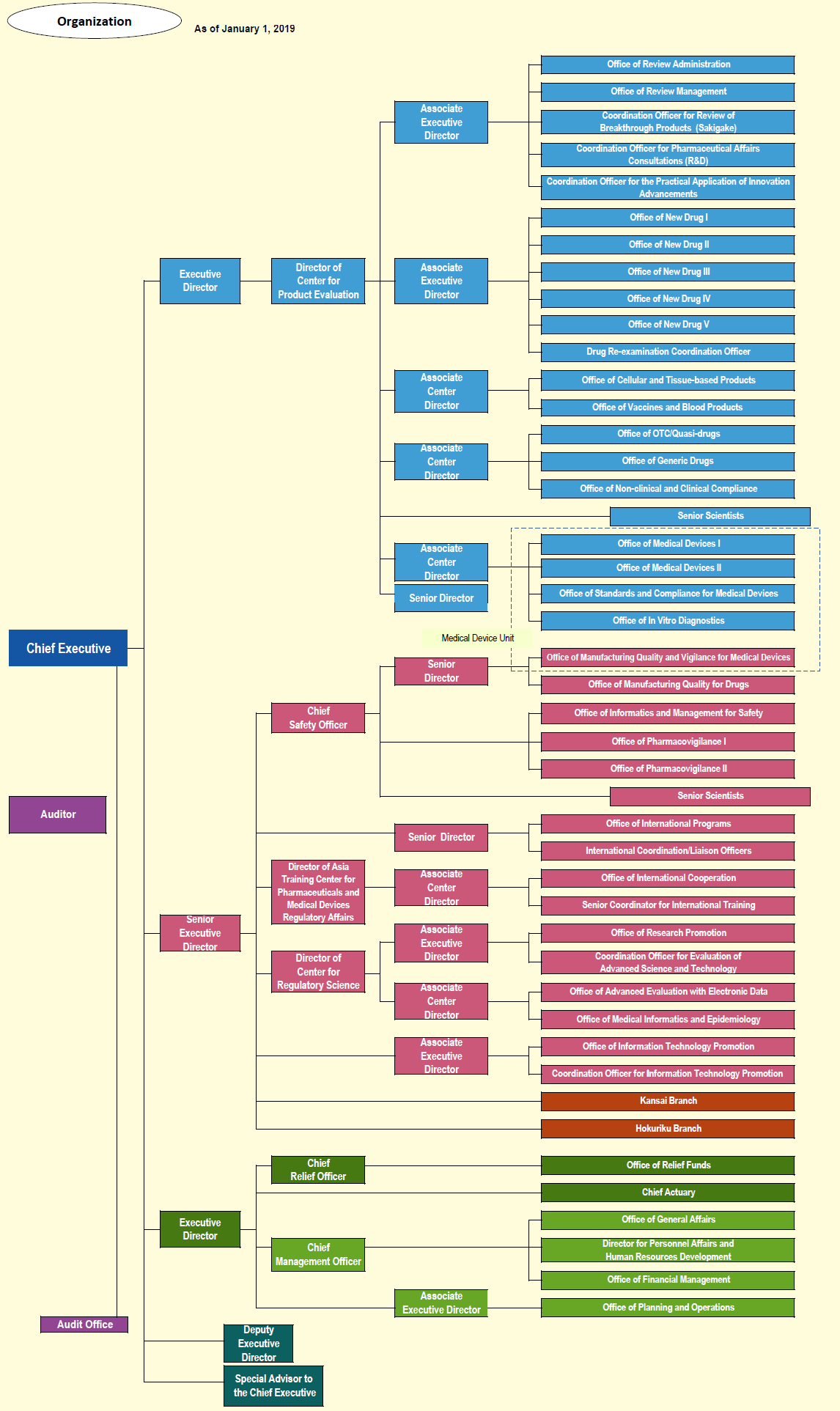
图3
二、PMDA 的主要业务
(一)审查
审查业务旨在管控风险、降低风险,是上市前对产品安全有效性的审核,审查业务包括临床试验和其他问题的咨询,对药品、医疗器械和再生医疗产品的合规性审查、再审查 / 再评价 , 针对按法规要求实施的试验的科学性、伦理性进行调查(GCP/GLP/GPSP 符合性评估), 生产过程和设施的 GMP/QMS/GCTP 检查 , 已注册认证机构的检查 , 标准的制修订等。
根据新版《药事法》,对初次获得批准的医疗器械,经一定时期后,要进行再审查。新设计的、结构新颖的或采用新原理的医疗器械,在获得初次批准后第四年,接受再审查。具有新效力、新用途或新性能的医疗器械,则在获得初次批准后第三年,接受再审查。
自 2015 年 10 月医疗器械审查部门实行新体制,由原来两个审查部调整为三个,分工见表 1。
表 1医疗器械审查部门分工

同时设有八个跨部门的小组,包括:
(1)临床评价小组;
(2)生物学安全小组;
(3)电气安全小组(含激光);
(4)软件小组(含网络安全应对);
(5)后发小组(包括合作计划:实质等同的明确化);
(6)国际应对小组,含IMDRF(International Medical Device Regulators Forum,国际医疗器械监管者论坛);
(7)监管科学小组(监管科学案例策划、与监管科学推进部的协调,以及对非其他小组管辖的监管科学案件的应对);
(8)再生医疗制品审查部、生物源器械办公室(生物源制品的安全性评价)。
(二)安全对策
安全对策业务是指上市后的安全措施,旨在持续性降低风险,是PMDA 与厚生省一同协作,为了保证医疗器械的安全、放心使用而实施。 PMDA与厚生省从制造商、经销商、医疗机构等处收集与医疗器械产品质量、有效性、安全性相关的信息,并对收集的信息进行科学的调查、探讨,形成的安全对应策略。根据各项规定要求,在 PMDA 官网上不仅可以查到审查相关的资料,同时可以查到紧急安全性信息、关于医疗安全信息的通知等。
(三)健康损害救济
健康损害救济旨在为医疗领域健康已受到的伤害采取救助措施,此业务与审评审批业务关系不大,因此本文未深入研究探讨。
三、PMDA 的承认审查
1. 新医疗器械:与已批准的医疗器械在结构组成、使用方法、效果及性能方面有明显差异的医疗器械。
2. 改良医疗器械:不属于新医疗器械或后发医疗器械的医疗器械。
3. 后发医疗器械:被认为与已批准的医疗器械在结构组成、使用方法、功能、效果及性能等有等同性的医疗器械,即与已批准医疗器械在构造、使用方法、效果及性能本质上等同的产品,申请认证或承认时不需要提供临床试验数据。
新医疗器械与改良医疗器械一般无相应的审查标准,无论风险等级为Ⅱ、Ⅲ还是Ⅳ级,均由 PMDA 进行审评,厚生省承认。自 2009 年起,对于已有审查标准的后发医疗器械,可由第三方认证机构认证;无审查标准的后发医疗器械仍由 PMDA 审评,厚生省承认。
日本医疗器械获得承认的流程见下图4:
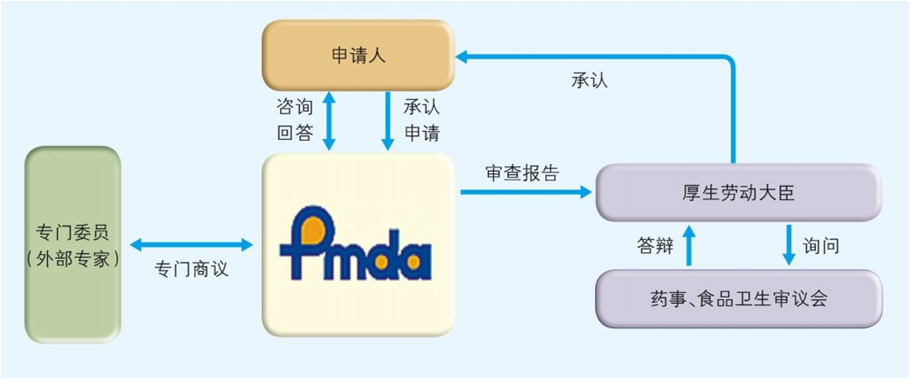
图4 医疗器械承认流程图
专门委员由 PMDA 从各学科中经验丰富者中选出并任命 , 名单在PMDA 网站公布。与专门委员商议的制度与我中心的专家咨询制度类似,有信函商议和会议商议两种方式。
往期回顾:如何处理日本(PMDA)医疗器械注册中的MAH制度(一)

