
 澳大利亚
澳大利亚 韩国
韩国 巴西
巴西 日本
日本 俄罗斯
俄罗斯 中国台湾
中国台湾 中国
中国 中国香港
中国香港 欧盟
欧盟 印度尼西亚
印度尼西亚 马来西亚
马来西亚 新加坡
新加坡 菲律宾
菲律宾 美国
美国 加拿大
加拿大 印度
印度 越南
越南 泰国
泰国 沙特阿拉伯
沙特阿拉伯如何处理日本(PMDA)医疗器械注册中的MAH制度(三)

本期主要讲日本法规中对MAH和DMAH的区别,以及中国企业进入日本市场选择MAH的策略。
从医疗器械市场容量来看,日本有着8倍俄罗斯、4倍韩国医疗器械市场的容量。由于日本本土制造业的人力成本和法规合规成本比较高,日本的医疗器械基本进口的量是出口量的两倍,所以日本市场可见一斑。
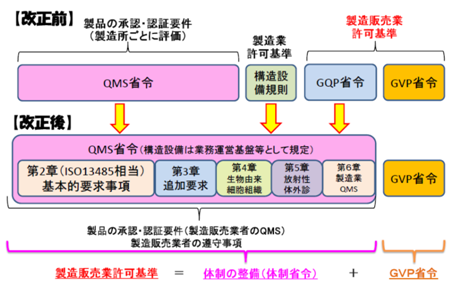
中国企业如果要去日本第一件很重要的一件事,就是选择MAH(Marketing Authorized Holder),因为在日本的2014年更新法《医药品医疗器械法》- PMD Act中,把原先的GQP取消,把日本对制造企业要求的QMS延申至MAH。换句话说,日本法规里的审核主体不在是以工厂为导向,而是以日本本土MAH为导向。
我们来看一段法规:
“製造販売業者への要求事項は、欧州代理人や米国代理人の要件よりも大幅に厳しく大きいものとなっています。製造販売業者は、日本に居所を有する必要があり、都道府県から正式な許可を受け、総括製造販売責任者、国内品質業務運営責任者、安全管理責任者など、資格要件を満たした人材を雇う必要があります。
また製造販売業者は厚生労働省令第169号の製造管理および品質管理の基準に関する省令(QMS省令)および厚生労働省令135号の製造販売後安全管理の基準に関する省令(GVP省令)に基づき、マネジメントシステムを構築し、実施、維持する必要があります。
日本に事業所を持たない製造業者は、その販売代理店に製造販売業者になってもらうかもしれませんが、その場合、販売代理店は製造販売業者として医療機器の製造販売承認といった製品登録を保有するため、当該医療機器とその製造販売を完全にコントロールすることになります。“
相关翻译:
“对制造分销商的要求比欧洲和美国代理商的要求严格得多。制造分销商必须在日本有固定住址,获得日本政府的正式许可,必须雇用符合资格要求的人员,例如制造和销售总经理,国内质量运营经理,安全经理等。
并且,制造分销商必须依据关于管理和质量控制标准的《厚生劳动省条例》第169号(QMS部长条例)以及关于售后安全管理标准的《厚生劳动部条例》第135号(GVP部长条例)来构建实施和维护管理系统(体系)。
在日本没有办事处的制造商,可以请销售代理商成为制造代理商,但是在这种情况下,制造代理商必须对该产品进行医疗器械注册,并拥有产品的制造贩卖授权,从而可以控制医疗器械及其生产和销售。”
按照法规要求,日本的MAH所持有的证书为制造贩卖业许可证,制造工厂施行厚生劳动省登入制。中国企业如果想在日本自己建立MAH分支,需要有相关的日本本土具有资质的人员,类似于我们中国的管代制和内审员制度。只有具备有相关资质的公司才能成为MAH。中国制造企业需要对在日本境内的公司进行了解,避免产生无效或缺乏有效资质企业成为自己的MAH。
外国特例承認制度と選任製造販売業者(DMAH)
“日本の厚生労働省は、クラスII、III、IVの医療機器の外国製造業者に、外国特例承認制度を設けることにより、当該医療機器の製造販売承認・製造販売認証を有することを許可しています。この外国特例承認制度により、外国製造業者は製造販売業者の代わりに、自らの名前で当該医療機器の製造販売承認・製造販売認証を取得することができ、外国製造業者は、みなし製造販売業者(製造販売業者等)となることができます。
日本厚生劳动省通过对II、III、IV等级医疗设备的外国制造商设立外国特别许可制度,允许其拥有该医疗设备的制造贩卖许可。根据外国特别许可制度,外国制造业者可以代替制造销售业者以自己的名字取得该医疗设备的制造贩卖许可及制造贩卖认证,外国制造业者可以成为制造贩卖业者(制造贩卖业者等)。
在这种情况下,有必要指定一个日本国内制造分销商(D-MAH)。DMAH有义务在日本执行基于QMS的质量管理,基于GVP的售后安全管理以及包括产品应用在内的各种其他业务。简单的说中国II、III、IV类医疗器械企业可以选择D-MAH在日本销售医疗器械,
往期回顾:如何处理日本(PMDA)医疗器械注册中的MAH制度(二)

