
 澳大利亚
澳大利亚 韩国
韩国 巴西
巴西 日本
日本 俄罗斯
俄罗斯 中国台湾
中国台湾 中国
中国 中国香港
中国香港 欧盟
欧盟 印度尼西亚
印度尼西亚 马来西亚
马来西亚 新加坡
新加坡 菲律宾
菲律宾 美国
美国 加拿大
加拿大 印度
印度 越南
越南 泰国
泰国 沙特阿拉伯
沙特阿拉伯如何将医疗器械注册成功进入日本市场:PMD Act审核程序及流程解析

如果企业希望把医疗器械产品投放到日本市场必须要满足日本的Pharmaceutical and Medical Device Act (PMD Act),以前我们知道是JPAL,这个法规是2014年药事法的改版。该法规规定绝大部分III类和IV类器械需要Pharmaceuticals and Medical Devices Agency (PMDA)/独立行政法人进行审核。目前也出版了一小部分英文的法规,这些法规可以在外面麦祥医药科技的网站里找到,但是语言问题和复杂的认证程序还是日本医疗器械注册的一个困难点。下面我们对不同的类别进行一个简述。
“Toroku”注册
在PMD Act的要求下,TOROKU注册系统要求国内的制造商必须向政府授权的当地的主管机关注册工厂信息,包括产品设计,生产,关键工序的信息;国外的制造商必须向MHLW注册制造商信息。
另外,依据产品的类别,分别按“Todokede, Ninsho and Shonin”程序进行医疗器械注册。
ClassI 器械:上市前提交Todokede
I类器械上市前必须由其MAH或DMAH向PMDA提交相关技术文件,这份文件不需要经过PMDA的审核和批准。
ClassII 器械:上市前认证Ninsho
作为特殊控制的II类器械上市前必须要经过上市前认证。认证机构(RCB)为PMDA授权可以进行PMDA认证的机构。
ClassII、III、IV 器械(以及创新器械):上市前批准Shonin
除了特殊控制的II类器械外的其他II类器械和III,IV类器械必须要由其MAH或DMAH像PMDA提交上市前批准的申请,并经过PMDA批准后才能注册他们的产品,并投放市场。
医疗器械必须要由其市场授权人MAH 或DMAH (Marketing Authorization Holder or Designated Marketing AuthorizationHolder)通过以下程序去注册其产品。
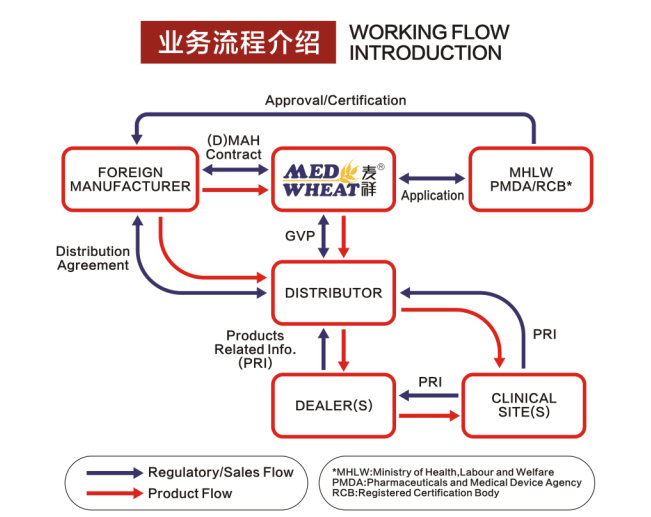
以下为我们麦祥的业务流程:
境外生产企业与麦祥及其日本境内的MAH签订合作协议之后,境外生产企业在麦祥日本的帮助下,由麦祥日本的MAH向PMDA独立行政法人/日本厚生劳动省/RCB监管机构递交相关注册资料。日本药监在适当时,对境外生产企业进行QMS审核,当体系和技术文件都被审核完毕且符合要求后,境外企业获得认证/承认。进入正常运营,境外生产商将医疗器械产品运往麦祥日本的MAH所在地,依据QMS进行检验通过后,方可委托境外企业商妥的经销商及零售商,之后可分别运往临床使用地。在之后的长时间运营中,境外企业持续符合产品品质要求,并符合QVP的法规监管。
有任何有关日本医疗器械市场的相关问题可通过下方图片中的垂询方式与我们联系:

